Introduction
Aliarcobacter butzleri (formerly Arcobacter butzleri) is a Gram-negative, aerotolerant, curved rod-shaped bacterium belonging to the Campylobacteraceae family (Müller et al., 2020). It is ubiquitously present in diverse environmental niches including surface water, wastewater, raw meats, and a range of foods such as poultry, beef, dairy products, seafood, pork, lamb, and vegetables (Chieffi et al., 2020; Hsu & Lee, 2015). As a result of its adaptability, Aliarcobacter butzleri (A. butzleri) participates in a dynamic ecological cycle involving food sources, animal reservoirs, and potential transmission to human hosts (Medina et al., 2022). Though often found as a commensal in livestock and poultry, its increasing detection in foodborne outbreaks and water samples raises public health concerns regarding its zoonotic potential (Ramees et al., 2017). Recent molecular epidemiology studies have highlighted the global prevalence of A. butzleri in food chains and aquatic systems, suggesting that it may be underdiagnosed due to limitations in detection methods (Ferreira et al., 2019). Genomic evidence also indicates the presence of multiple virulence and resistance determinants, supporting its classification as an emerging foodborne and waterborne pathogen (Fanelli et al., 2019).
In humans, A. butzleri is associated with gastrointestinal disorders such as acute enteritis, watery diarrhea, abdominal cramps, and, less frequently, bacteremia—particularly in immunocompromised patients. Animal infections include mastitis in cattle and reproductive disorders in pigs and sheep (Chieffi et al., 2020; Collado & Figueras, 2011). Although large-scale outbreaks remain rare, likely due to misidentification or inadequate diagnostic testing, sporadic cases are increasingly reported worldwide (Novak et al., 2024). Transmission of A. butzleri typically occurs via consumption of contaminated food
or water in humans and animals (Chieffi et al., 2020), although direct contact with animals or contaminated surfaces is also another possible way to enter into the human body (Chieffi et al., 2020; Giacometti et al., 2015). The clinical significance of A. butzleri is further exacerbated by its rising multidrug resistance (MDR) profile. Several isolates have demonstrated resistance to common antibiotics including fluoroquinolones, erythromycin, ciprofloxacin, tetracycline, clindamycin, florfenicol, and nalidixic acid (Abay et al., 2012; Müller et al., 2020; Oliveira et al., 2023). Additionally, the organism’s intrinsic ability to produce biofilms enhances survival in hostile environments and may complicate eradication efforts. Moreover, in vitro analyses have confirmed that nearly all A. butzleri isolates can form robust biofilms under laboratory conditions (Oliveira et al., 2023). This combination of MDR and biofilm-forming capacity presents challenges in eradication and infection control. Despite these concerns, current understanding of its pathogenesis, resistance mechanisms, and potential therapeutic or diagnostic targets remains limited. While virulence-associated genes such as hecA, hecB, and irgA have been identified (Kietsiri et al., 2021), a holistic understanding of its functional genome is lacking. In addition, limited information exists regarding A. butzleri’s heavy metal resistance (Fanelli et al., 2019). While A. butzleri remains underexplored compared to other enteric pathogens, a few studies have suggested putative therapeutic targets, such as outer membrane proteins, efflux pumps, and resistance genes associated with fluoroquinolone and β-lactam resistance (Fanelli et al., 2019; Ma et al., 2022; Mateus et al., 2021). However, systematic identification and validation of essential, non-homologous proteins as therapeutic candidates are still lacking, especially using integrative functional genomics approaches. This highlights the need for deeper investigation into its hypothetical proteins, many of which may play critical roles in survival and pathogenesis.
With the availability of complete genome sequences for A. butzleri, many of its proteins remain annotated as “hypothetical,” with no experimentally verified functions. In bacteria, more than 30% of encoded proteins are typically uncharacterized and termed hypothetical proteins (HPs) (Shahbaaz et al., 2015). These HPs are not simply genomic noise—they may participate in key processes such as host adhesion, toxin secretion, stress tolerance, or antibiotic resistance (Naveed et al., 2022). Functional annotation of these proteins is thus essential to unravel novel virulence mechanisms or therapeutic targets. Advances in bioinformatics now allow the functional prediction of HPs through integrative analyses of sequence homology, conserved domains, subcellular localization, structural modeling, and interactome mapping (Pranavathiyani et al., 2020). These approaches have been successfully applied in various bacterial pathogens such as Borrelia burgdorferi (Khan et al., 2016), Chlamydia trachomatis (Turab Naqvi et al., 2017), Helicobacter pylori (Naqvi et al., 2016), Haemophilus influenzae (Shahbaaz et al., 2013), and Mycobacterium tuberculosis.(Yang et al., 2019). However, to date, no systematic computational effort has been undertaken to characterize the HPs in A. butzleri strain BNI-3166.
To fill this gap, the present study employed a comprehensive computational pipeline to functionally annotate 356 HPs from A. butzleri BNI-3166. We prioritized proteins based on essentiality, non-homology to the human host, presence of conserved functional domains, and potential roles in virulence or druggability. This integrated approach enabled us to identify promising therapeutic and prophylactic targets for future experimental validation. Our findings provide a foundation for understanding the molecular underpinnings of A. butzleri pathogenicity and support the development of targeted antimicrobial interventions.
Materials and methods
Sequence retrieval
The complete proteome of A. butzleri strain BNI-3166 (NZ_CP184264.1) was downloaded from the National Center for Biotechnology Information (NCBI), a widely used platform for genomic and biomedical resources (https://www.ncbi.nlm.nih.gov/). All the HPs in fasta format were extracted from the downloaded proteome data using a Python script.
Essentiality and non-homology analysis
The HPs identified from A. butzleri BNI-3166 strain were screened for essentiality using a protein BLAST (BLASTp) search against the Database of Essential Genes (DEG 15.2 (Luo et al., 2021)), which includes experimentally validated essential genes from various bacterial, archaeal, and eukaryotic species. The parameters used for this similarity search included an e-value threshold of ≤ 0.0001 and a bit-score of ≥ 60. Proteins that showed significant similarity to entries in the DEG database were considered as potentially essential. Following this, a BLASTp (Altschul et al., 1990) comparison was performed between the identified essential proteins and the human proteome to eliminate homologous proteins. We considered proteins with no significant matches (e-value ≥ 0.0001) to any human proteins as non-homologous and pathogen-specific. These essential non-homologous (ENH) proteins were then selected for subsequent analyses.
Function prediction
Several widely accepted bioinformatics tools and databases was used to functionally annotate the ENH proteins of A. butzleri BNI-3166 strain identified in the previous analysis. These include Pfam, InterPro, CATH, SUPERFAMILY, SMART, SCANPROSITE, CDD-BLAST, MOTIF, and PANTHER. These tools help in identifying conserved domains and predicting the possible function of proteins. Pfam 37.4 (Mistry et al., 2021), InterPro 106.0 (Blum et al., 2025), and SUPERFAMILY 2 (Gough et al., 2001) were used to predict protein functions based on sequence similarity and conserved domain matches. SMART v9 (Letunic et al., 2021) and CATH v4.4 (Sillitoe et al., 2015) assisted in analyzing the domain architecture and classifying domains within a structural hierarchy. The Conserved Domain Database (CDD 3.21) (Lu et al., 2020) was employed to detect known conserved domains in the protein sequences. In addition, the MOTIF (https://www.genome.jp/tools/motif/) (Henikoff et al., 2000; Kanehisa, 1997) tool was used to identify short conserved patterns or motifs, and the PANTHER 19 (Mi et al., 2019) database was used to classify proteins into families and predict their functions based on evolutionary relationships. To ensure reliability, we selected only those HPs that showed functional domains presence in at least three of the tools for further downstream analysis.
Homology search
To manually annotate the selected HPs, their FASTA sequences were used in a BLASTp (Altschul et al., 1990) search against the NCBI non-redundant protein database. Only those hits with a sequence identity of 90% or higher were considered as a best hit for those HPs.
Physicochemical characterization
The ProtParam (Gasteiger et al., 2005) tool, available through the ExPASy server, to assess the physicochemical properties of the selected HPs. This analysis provided important information, including the molecular weight, isoelectric point (pI), extinction coefficient, instability index, aliphatic index, and GRAVY (Grand Average of Hydropathicity) score, all of which offer insights into the structural stability, solubility, and overall behavior of the proteins.
Promoter analysis
Promoter regions of the selected HPs were analyzed using the BPROM (http://softberry.com) tool with default settings. The corresponding DNA sequences were retrieved from the NCBI database. In this analysis, we manually assigned the Shine-Dalgarno (SD) sequence.
Protein–protein interaction analysis
To predict the interaction of the selected proteins, the STRING v12 database was used with a confidence score threshold set above 0.7 to ensure reliable results. Since the BNI-3166 strain was not available in the database, Arcobacter butzleri RM4018 was used as the reference genome (Szklarczyk et al., 2019). Both physical and functional interactions were considered in building the networks. The resulting interaction networks were then visualized using Cytoscape software v3.10.3 (Shannon et al., 2003).
Gene ontology analysis
The gene ontology (GO) terms for all the selected HPs were predicted using the Argot2.5 v2.5 online tool, which helps assign functional annotations based on available data (Lavezzo et al., 2016).
Virulence prediction
To identify virulent proteins among the selected HPs, a BLASTp search against the core dataset of the Virulence Factor Database (VFDB) was performed using a cut-off e-value of ≤ 0.01. VFDB (Chen et al., 2016) is a specialized resource containing experimentally validated bacterial virulence factors. Additionally, the selected HPs were analyzed using VICMpred (Saha & Raghava, 2006), an SVM-based tool that classifies Gram-negative bacterial proteins based on their amino acid composition into functional categories. Another tool, VirulentPred 2.0 (Garg & Gupta, 2008), was also used to predict virulence. If at least two out of these three tools predicted a HP as virulent, it was considered a putative virulence factor in this study.
Druggability and subcellular localization analysis
To evaluate the druggability of the annotated HPs, similarity search against the ChEMBL database was performed using a cut-off e-value of ≤ 0.00001. ChEMBL is a curated database that contains information about bioactive drug-like compounds and their experimentally tested targets (Gaulton et al., 2012). To predict where each protein is located within the cell, two tools, PSORTb v3.0.3 (N. Y. Yu et al., 2010), and CELLO v2.5 (C. Yu et al., 2004), were used. PSORTb combines experimental data with computational predictions, while CELLO uses a support vector machine (SVM)-based system for classification. Additionally, to identify transmembrane regions within the proteins, SOSUI v1.11 (Hirokawa et al., 1998), HMMTOP v2.0 (Tusnády & Simon, 2001), and TMHMM v2.0 (Krogh et al., 2001) tools were applied. Predicting protein localization and membrane-spanning regions helps in understanding their possible roles and suitability as drug targets and possible vaccine candidates.
Structure prediction
Understanding the 3D structure of a protein is important for revealing its molecular function and for designing structure-based drugs. In this study, the 3D structure of the selected HPs was predicted using the AlphaFold v3.0.1 server (John et al., 2021). The quality and reliability of the predicted structure were then validated using the SAVES server (https://saves.mbi.ucla.edu/).
Ligand binding site prediction
The PrankWeb server (https://prankweb.cz/) was used to predict potential ligand binding sites in the AlphaFold3-modeled structures of the selected drug target proteins. This tool employs a machine learning–based pocket prediction algorithm to identify ligand-accessible surface cavities and provide confidence scores based on residue environment and conservation. Each uploaded PDB file was analyzed under default parameters (Jendele et al., 2019).
Antigenicity prediction
The antigenicity potential of the two vaccine candidate proteins was evaluated using VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html), which predicts probable antigens based on their physicochemical properties without alignment. The threshold was set to the default value of 0.4 for bacterial models. Proteins scoring above this threshold were considered likely antigenic, while those below were considered non-antigenic (Doytchinova & Flower, 2007).
Results and discussion
In this study, HPs from the A. butzleri BNI-3166 genome were analyzed using various in-silico tools to identify potential candidates for drug and vaccine development. The overall workflow followed in this analysis is shown in Figure 1.
Figure 1. Overview of the workflow followed in this study.
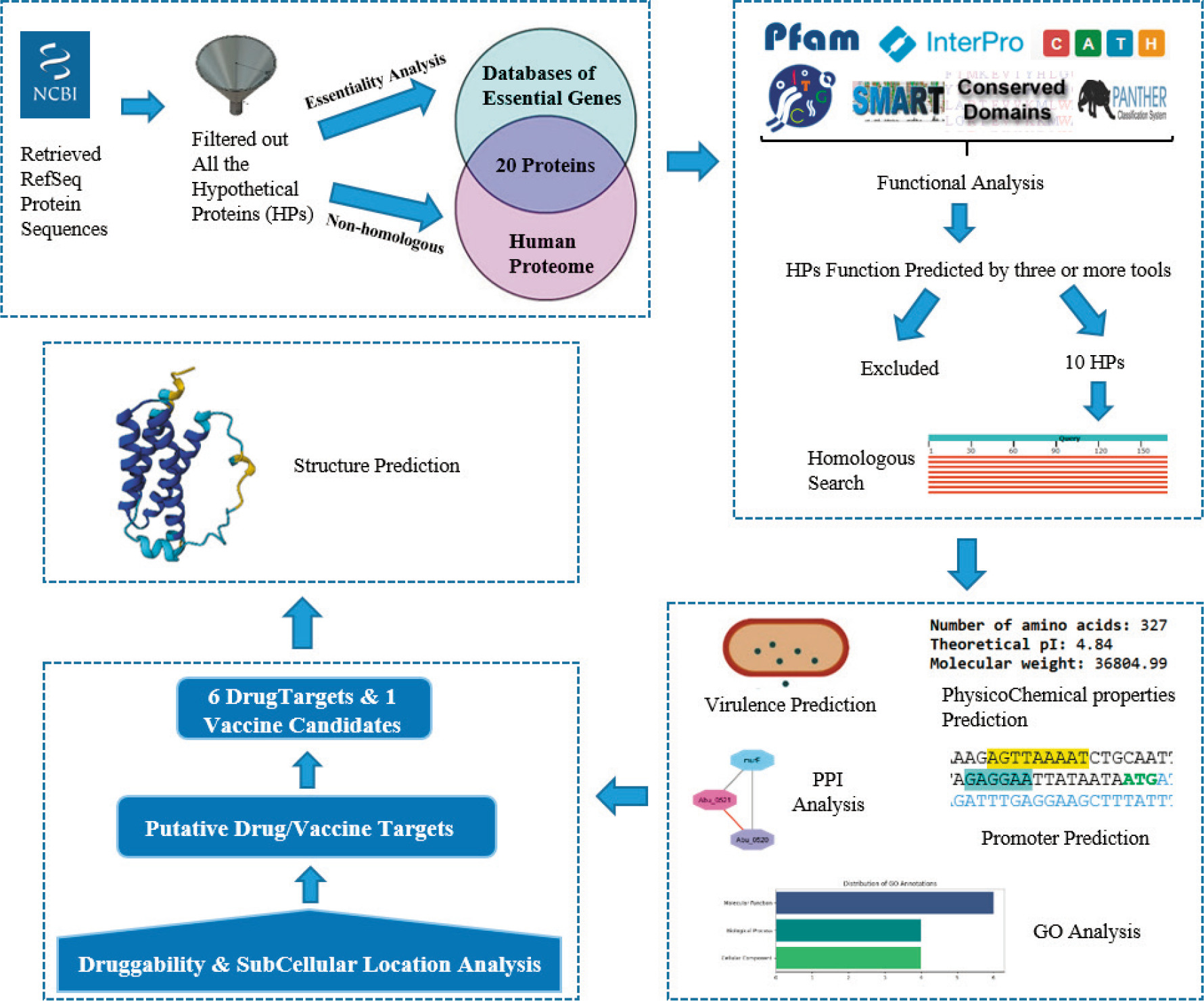
Identification of essential pathogen-specific proteins
The complete genome of A. butzleri BNI-3166 consists of a single circular chromosome approximately 2.4 Mb in size and encodes a total of 2,367 protein-coding genes. All protein sequences were retrieved for this study. Among these, 356 proteins were identified as HPs based on the absence of functional annotation (Supplementary File 1). To identify potential therapeutic targets, these 356 HPs were subjected to essentiality and non-homology analysis. The goal of the essentiality analysis was to find proteins that are vital for the pathogen’s survival, as targeting such proteins may disrupt crucial biological processes and be lethal to the organism. In parallel, non-homology analysis was performed to exclude proteins that share similarity with human proteins, ensuring that the shortlisted targets are specific to the pathogen and would minimize the risk of adverse effects in the host. For essentiality screening, a BLASTp search was conducted against the Database of Essential Genes (DEG), which contains experimentally verified essential proteins from various bacterial species. Out of the 356 HPs, 20 proteins showed significant similarity to known essential bacterial proteins and were considered putative essential proteins in A. butzleri. These 20 proteins were then compared to the human proteome using BLASTp to assess homology. None of them showed any significant similarity to human proteins, confirming their pathogen-specific nature. These 20 ENH proteins (listed in Supplementary File 2) are both critical to the survival of A. butzleri and absent in the human host, making them promising candidates for drug or vaccine development. Further analyses, including functional annotation, physicochemical profiling, virulence prediction, druggability assessment, and subcellular localization, were carried out to refine and prioritize these potential therapeutic targets.
Functional analysis
To determine the possible roles of the 20 essential non-homologous hypothetical proteins, we performed domain-based functional analysis, as domains are key structural, functional, and evolutionary units of proteins. Each protein sequence was analyzed using multiple bioinformatics tools, including Pfam, InterPro, CATH, SUPERFAMILY, MOTIF, SMART, PANTHER, and CDD-BLAST (Supplementary File 3). Based on the outputs from these tools, functions were confidently assigned to 10 HPs that showed consistent domain predictions in at least three or more programs (Table 1). These proteins were considered to have reliable functional annotations. For 7 other HPs, domains were detected by fewer than three tools, suggesting the need for further investigation. The remaining 3 HPs showed no detectable domains or significant similarity in any of the databases used. Among the 10 functionally annotated proteins, most were categorized as enzymes, binding proteins, transporters, or regulatory proteins (Figure 2). Predicted molecular functions for the 10 annotated HPs are discussed in detail within subsequent functional categories.
Table 1. Functional annotation of hypothetical proteins identified in A. butzleri strain BNI-3166.
| No. | HP Accession Id | Predicted Function | |
|---|---|---|---|
| 1 | WP_004510585.1 | Tetratricopeptide repeat | |
| 2 | WP_004510999.1 | WD40 repeat-like | |
| 3 | WP_012012715.1 | Cell division protein FtsX-like permease | |
| 4 | WP_020848489.1 | Actin-like ATPase domain | |
| 5 | WP_041644687.1 | ThiD2 family | |
| 6 | WP_193220824.1 | Chemotaxis phosphatase CheZ | |
| 7 | WP_260901056.1 | membrane-associated subunit of NADH:ubiquinone oxidoreductase complex | |
| 8 | WP_ | 260902335.1 | Uncharacterized conserved protein YgiM, contains N-terminal SH3 domain |
| 9 | WP_419970369.1 | Dolichyl-phosphate-mannose-protein mannosyltransferase | |
| 10 | WP_420061029.1 | D-alanine-D-alanine ligase | |
Figure 2. Predicted functional classification of 10 hypothetical proteins from A. butzleri.
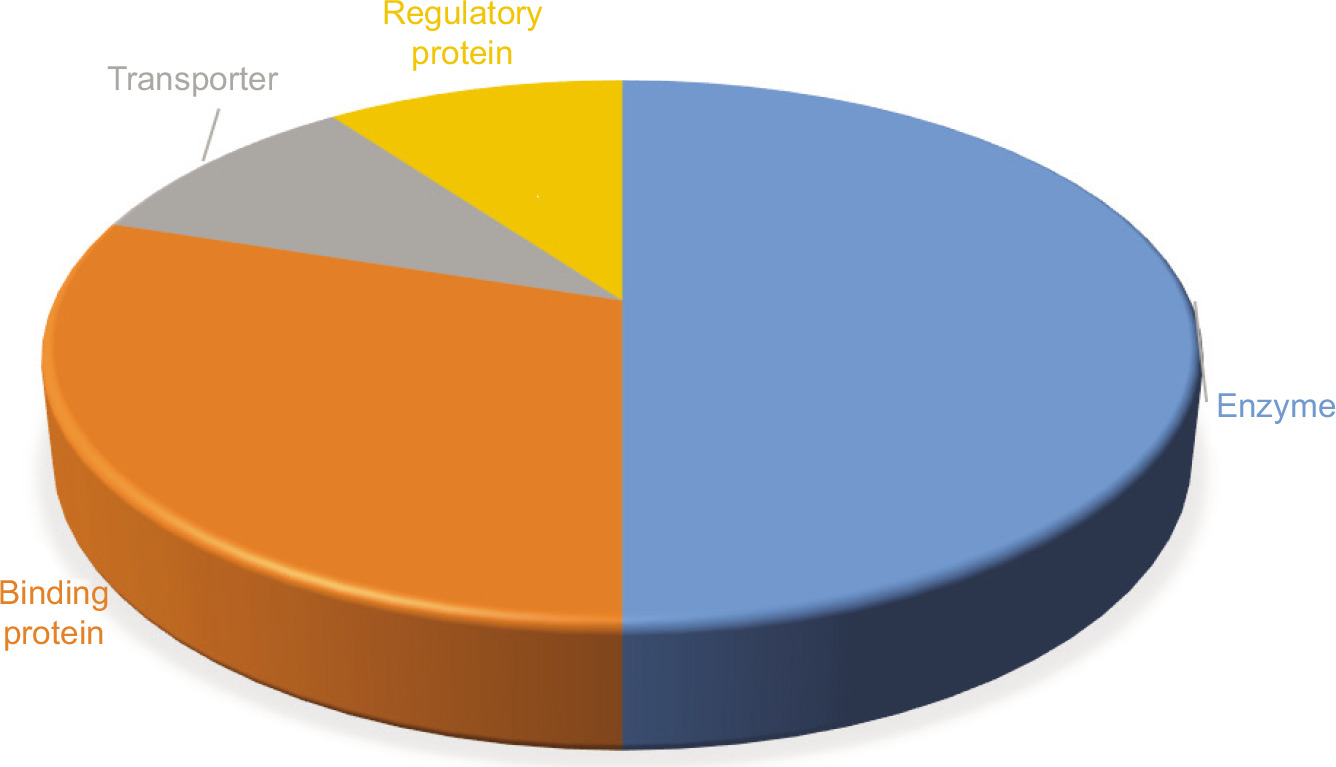
Enzymes
Enzymes serve as biological catalysts, playing pivotal roles in biochemical pathways and metabolic processes across all organisms. In pathogens, they are especially vital for nutrient utilization, survival, growth, and host-pathogen interactions (Bjornson, 1984). Among the 10 functionally annotated ENH proteins of A. butzleri BNI-3166, five were identified as enzymes belonging to distinct functional classes, each playing a vital role in bacterial metabolism and survival. The protein WP_041644687.1 was predicted to be part of the ThiD2 family, involved in strictly monofunctional hydroxymethylpyrimidine monophosphate (HMP-P) kinases, which catalyze the phosphorylation of HMP-P to HMP-PP in the thiamine biosynthesis pathway. Unlike canonical ThiD kinases, ThiD2 proteins exhibit high substrate specificity, phosphorylating only HMP-P and not free HMP or structurally similar analogs, thereby avoiding incorporation of potentially toxic compounds into the pathway (Thamm et al., 2017). WP_193220824.1 showed similarity to chemotaxis phosphatase CheZ, known to regulate bacterial motility by dephosphorylating the response regulator CheY, a key component of chemotactic signaling (Zhao et al., 2002). The protein WP_260901056.1 matched a membrane-associated subunit of the NADH:ubiquinone oxidoreductase (Complex I), essential for electron transport and proton translocation, contributing to energy generation in bacteria (Casutt et al., 2011). WP_419970369.1 was identified as a dolichyl-phosphate-mannose-protein mannosyltransferase, an enzyme involved in protein glycosylation, which supports bacterial viability by stabilizing proteins (Lommel & Strahl, 2009) and modulating host-pathogen interactions (Borong et al., 2020). Lastly, WP_420061029.1 was predicted as D-alanine–D-alanine ligase (Ddl), a key enzyme in peptidoglycan synthesis, forming the D-Ala–D-Ala dipeptide necessary for bacterial cell wall integrity. Numerous studies have validated Ddl as a promising antibacterial target because it lacks a human counterpart (Liu et al., 2006). Notably, 5 HPs were identified as enzymes, and these were further classified into functional subcategories such as kinases, phosphatases, ligases, transferases, and oxidoreductases (Table 2).
Table 2. Sub-classification of identified enzymes.
| No. | HP Accession Id | Sub-category |
|---|---|---|
| 1 | WP_041644687.1 | Kinases |
| 2 | WP_193220824.1 | Phosphatases |
| 3 | WP_260901056.1 | Oxidoreductases |
| 4 | WP_419970369.1 | Transferases |
| 5 | WP_420061029.1 | Ligases |
Binding proteins
Three of the ENH proteins identified from A. butzleri BNI-3166 are predicted to function as binding proteins, each featuring a distinct protein–protein interaction domain. The protein WP_004510585.1 contains a tetratricopeptide repeat (TPR) motif, a well-established alpha-helical scaffold that mediates assembly of multiprotein complexes and is implicated in bacterial pathogenic processes (Blatch & Lässle, 1999). Meanwhile, WP_004510999.1 exhibits a WD40-like repeat architecture, forming a β-propeller structure that typically serves as a stable platform for transient protein interactions and signal transduction in both eukaryotes and bacteria (Hu et al., 2017). Lastly, WP_260902335.1 is annotated as an uncharacterized conserved protein homologous to YgiM, harboring an N-terminal SH3 domain (COG3103), a domain commonly involved in protein recognition and regulatory processes in prokaryotic systems.
Transporter
The protein WP_012012715.1 was predicted to be a cell division protein FtsX–like permease, placing it within the ABC transporter family and classifying it as a transporter. FtsX, via its permease domain, acts as a membrane anchor linking cytoplasmic proteins like FtsZ (which forms the Z-ring at the division site) to periplasmic enzymes, coordinating the constriction of the division ring with cell wall remodeling (Alcorlo et al., 2020).
Regulatory protein
The protein WP_020848489.1 was annotated with an actin-like ATPase domain, classifying it as a regulatory protein. In certain ATPases, such as the plasma membrane calcium ATPase (PMCA), direct interaction with actin (which contains the actin-like ATPase domain) stimulates enzymatic activity. Actin oligomers can increase the affinity of PMCA for Ca2+ and enhance the rate of enzyme phosphorylation, leading to increased pump activity. This demonstrates a regulatory mechanism where actin dynamics directly influence the function of other ATPases (Dalghi et al., 2013).
Homology search, promoter region identification, protein–protein interaction, and gene ontology (GO) analysis
Out of the 10 selected HPs analyzed through BLASTp, several showed strong homology to proteins with known functional roles. These proteins (n = 4) were matched with high sequence identity to characterized proteins across different A. butzleri strains and related species, supporting the functional predictions made during domain analysis. The remaining proteins aligned with either uncharacterized conserved proteins or remained annotated as HPs. Overall, the BLASTp results were consistent with the earlier predictions and provided additional evidence for the putative roles of these HPs in A. butzleri (Supplementary File 4).
Promoter region analysis was conducted for all 10 selected HPs to identify regulatory elements involved in transcription initiation. Promoter sequences typically contain conserved motifs such as the Pribnow box (-10 region), the -35 box, and the Shine-Dalgarno (SD) sequence, which are essential for RNA polymerase binding and initiation of translation (Pribnow, 1975). Among the 10 proteins, only two, WP_004510585.1 and WP_012012715.1 had corresponding gene sequences available in the NCBI database. For these two proteins, promoter elements including the -10 and -35 regions and SD sequences were successfully predicted (Supplementary Figure 1). However, for the remaining proteins, gene sequences were unavailable in the NCBI database, and therefore their promoter regions could not be analyzed.
To explore the potential interaction partners of the selected HPs, a protein–protein interaction (PPI) analysis was carried out. The results revealed that WP_012012715.1 (predicted as cell division protein FtsX) showed a strong interaction with FtsE (interaction score: 0.811), a known partner in the bacterial cell division process. Another protein, WP_020848489.1, was found to interact with Gcp (score: 0.945), which is associated with glycoprotease activity (Miller et al., 2007). Additionally, WP_260901056.1 showed a high-confidence interaction with NuoCD (score: 0.999), a component of the NADH-quinone oxidoreductase complex involved in cellular respiration. These interaction results support the earlier functional predictions and provide further insights into the biological roles of these HPs (Supplementary File 5 and Supplementary Figure 2).
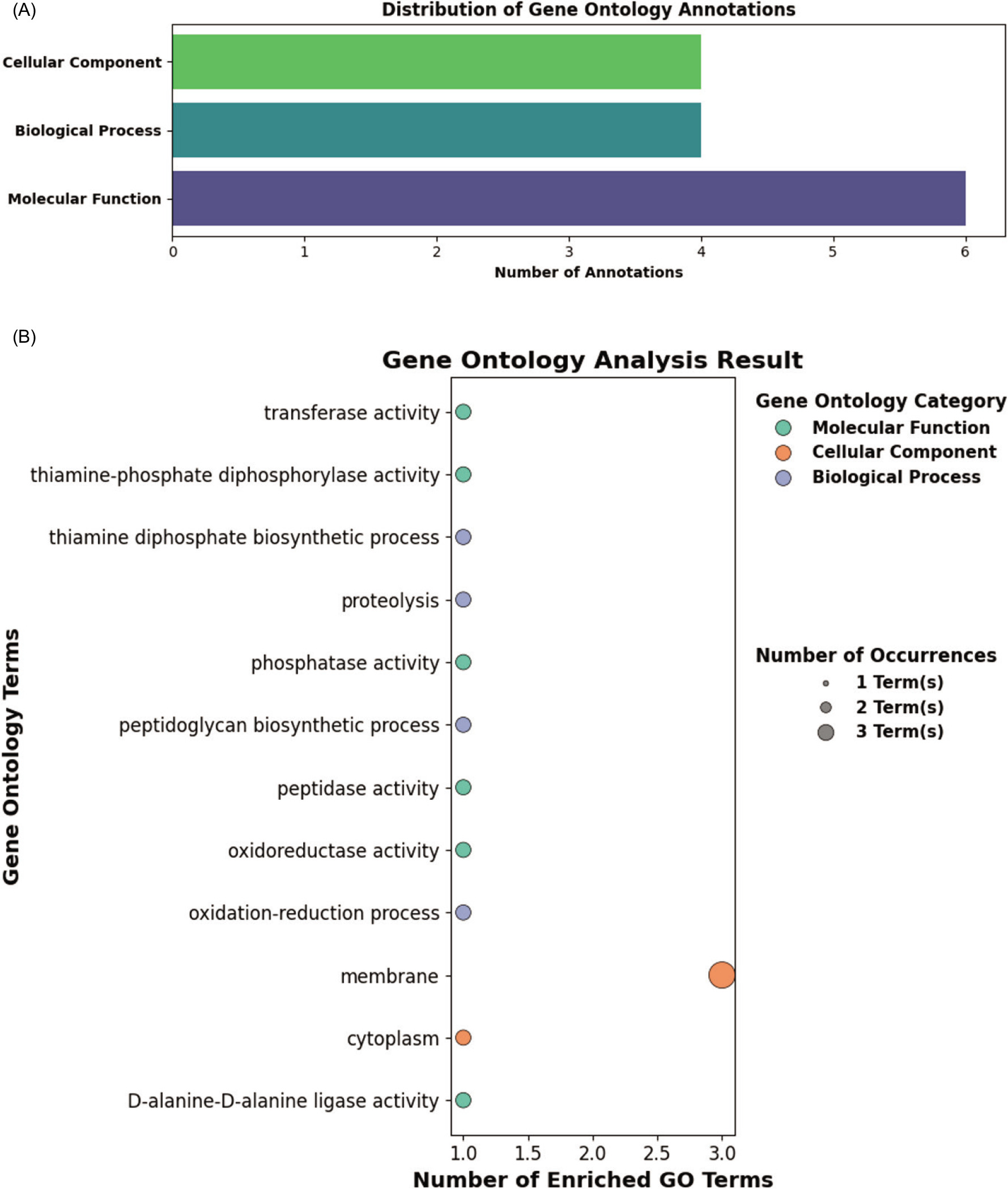
Gene Ontology (GO) terms for the selected HPs were predicted using the Argot2.5 server based on confidence scores (Supplementary File 6). Among the three main GO categories, the majority of terms fell under molecular function, while biological process and cellular component categories had an equal number of classifications (Figure 3a). Within the cellular component category, two distinct GO terms were identified, with three proteins predicted to be membrane-associated. Although membrane proteins can be challenging to study, they are known to play essential roles in bacterial physiology, especially in interactions with environmental stressors (Desvaux et al., 2006; Walian et al., 2012). Under the biological process category, four functions were identified: proteolysis, thiamine diphosphate biosynthesis, oxidation–reduction process, and peptidoglycan biosynthesis. For the molecular function category, six GO terms were observed, including D-alanine–D-alanine ligase activity, transferase activity, and oxidoreductase activity, among others (Figure 3b). These findings further support the functional roles predicted for the HPs.
Figure 3. Gene Ontology (GO) analysis of the 10 hypothetical proteins. (a) Distribution of the HPs across the three main GO categories. (b) Graphical representation of the associated GO terms.
Physicochemical properties and virulence factor prediction
The physicochemical features of the 10 annotated HPs were analyzed to better understand their biochemical behavior and potential for further experimental study. The molecular weights of the proteins ranged from 16.06 kDa to 47.88 kDa. Theoretical isoelectric points (pI) varied widely, with the lowest being 4.38 and the highest 9.33, which can help guide protein purification strategies. Protein stability was assessed using the instability index, where a value below 40 indicates a stable protein (Gill SC, 1989). In this study, 7 out of 10 proteins were predicted to be stable based on this criterion. The aliphatic index, which relates to protein thermostability, ranged from 86.21 to 150.89, suggesting good stability for most proteins. Hydropathicity was measured using the GRAVY score, which ranged from −0.729 to +0.719, with an average of −0.1192, indicating that most of the proteins are likely hydrophilic in nature. These calculated parameters are summarized in Supplementary File 7 and may be useful for designing future experimental work such as crystallization or drug-binding assays.
To identify potential virulence-related proteins among the annotated HPs, a homology search was performed against the core protein sequences dataset of the Virulence Factor Database (VFDB). This analysis revealed three proteins—WP_004510585.1, WP_193220824.1, and WP_260901056.1—with notable sequence similarity to known virulence factors from Legionella pneumophila, and Mycoplasma hyopneumoniae. Specifically, WP_004510585.1 and WP_193220824.1 shared sequence similarity with Dot/Icm T4SS secreted proteins, which are part of the Type IV secretion system in L. pneumophila, a mechanism known to translocate virulence effectors into host cells. While their presence in A. butzleri is suggestive, further investigation is needed to confirm similar roles. These systems play a crucial role in pathogenesis and are widely recognized as major determinants of bacterial virulence (Segal et al., 1999). Additionally, WP_260901056.1 showed high similarity to the P97/P102 adhesin complex found in Mycoplasma hyopneumoniae 232, which is involved in the adherence of the bacterium to host epithelial cells, facilitating colonization and infection (Raymond et al., 2018). The annotated HPs were further analyzed using the VICMpred server, which categorizes bacterial proteins into four classes: virulence factors, information molecules, cellular process proteins, and metabolism-related proteins. Based on this prediction, six proteins were associated with cellular processes, three proteins were linked to metabolic functions, and one protein was categorized under information storage and processing. Moreover, the VirulentPred 2.0 server predicted that nine out of ten proteins were likely virulent, while only one was classified as non-virulent. The three proteins (WP_004510585.1, WP_193220824.1, and WP_260901056.1) that were consistently identified as virulence-related by VFDB and VirulentPred 2.0 were considered putative virulence-related candidates in this study. These findings provide computational evidence supporting the potential involvement of these HPs in the pathogenicity of A. butzleri BNI-3166, though experimental confirmation is required. Table 3 summarizes the key findings of the analysis.
Table 3. Prediction of virulence potential in all 10 hypothetical proteins.
| No. | HP Accession Id | VICMPred | VFDB | VirulentPred 2.0 | |
|---|---|---|---|---|---|
| 1 | WP_004510585.1 | Cellular process | Virulent | Virulent | |
| 2 | WP_004510999.1 | Cellular process | No Result | Virulent | |
| 3 | WP_012012715.1 | Metabolism Molecule | No Result | Virulent | |
| 4 | WP_020848489.1 | Cellular process | No Result | Virulent | |
| 5 | WP_041644687.1 | Information and storage | No Result | Virulent | |
| 6 | WP_193220824.1 | Cellular process | Virulent | Virulent | |
| 7 | WP_260901056.1 | Metabolism Molecule | Virulent | Virulent | |
| 8 | WP_ | 260902335.1 | Cellular process | No Result | Virulent |
| 9 | WP_419970369.1 | Metabolism Molecule | No Result | Virulent | |
| 10 | WP_420061029.1 | Cellular process | No Result | Non-virulent | |
Druggability and subcellular localization analysis
The ChEMBL database was used to assess the druggability of the selected HPs. Out of the ten proteins analyzed, nine showed no similarity to any known drug targets listed in ChEMBL. Only one protein (WP_420061029.1) matched with three known targets, suggesting possible drug-related interactions. Based on these results, the nine unmatched proteins may represent potential novel drug targets, which could be further explored and validated through experimental studies.
The cellular localization of proteins is vital for their biological functions in a specific environment (C.-S. Yu et al., 2006). Subcellular localization and transmembrane helix predictions were performed for 10 HPs using PSORTb, CELLO, SOSUI, HMMTOP, and TMHMM. Six proteins were consistently predicted as cytoplasmic or soluble with no transmembrane helices, suggesting intracellular functions. Several cytoplasmic proteins play crucial roles in biosynthesis, transport, and regulation, helping environmental bacteria adapt and compete more effectively with other organisms in the same ecological niche (Nakashima & Nishikawa, 1994). Four proteins showed membrane-associated features, with predictions indicating localization to the inner or outer membrane and the presence of 1 to 10 transmembrane helices, indicating roles as potential transporters or membrane-bound enzymes (Supplementary File 8).
Predicted subcellular localization helped us categorize the proteins accordingly. Proteins present in the cytoplasm, periplasm, and inner membrane were considered potential drug targets, while those located in the outer membrane or extracellular region were classified as possible vaccine candidates (Barh et al., 2011). Based on this approach, we identified eight proteins as putative drug targets and two as possible vaccine candidates. These computational predictions provide a useful foundation for future experimental validation and therapeutic development efforts.
Tertiary structure predictions and validation
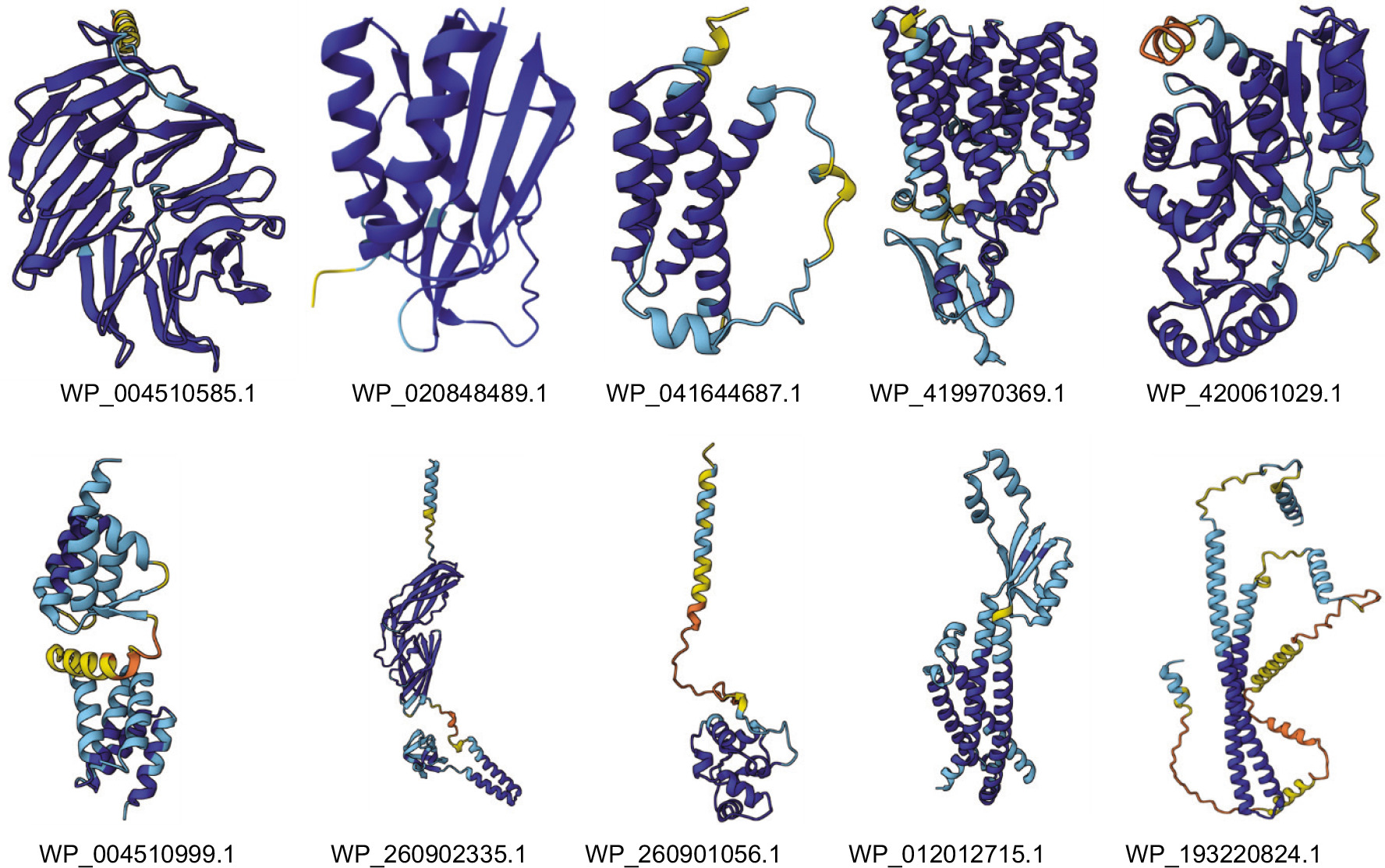
To better understand the structural features and functional domains of the selected proteins, tertiary structure modeling was performed using the AlphaFold3 server, as suitable template structures were not available for homology modeling. All 10 HPs were successfully modeled (Figure 4). The predicted structures were evaluated using the predicted TM-score (pTM), which measures how closely a model resembles a true protein structure. The pTM scores ranged from 0.44 to 0.92 (Supplementary File 9), where scores closer to 1.0 indicate higher confidence in the structural prediction. Model validation was performed using the SAVES server, and the resulting Ramachandran plots and scores, indicative of model quality, are provided in Supplementary Figure 3. These structural models provide a foundation for future studies, including structure-based drug design.
Figure 4. Predicted tertiary structures of all 10 hypothetical proteins. The pLDDT (Predicted Local Distance Difference Test) score, a per-residue confidence metric for predicted protein structures, is represented by a color scale where blue indicates very high confidence (pLDDT > 90), cyan signifies high confidence (90 > pLDDT > 70), yellow-orange denotes low confidence (70 > pLDDT > 50), and orange-red marks very low confidence (pLDDT < 50).
Ligand binding site identification
To assess the druggability of the eight previously identified hypothetical protein targets, we predicted their ligand-binding pockets using PrankWeb. Of the eight, six proteins (WP_004510585.1, WP_020848489.1, WP_041644687.1, WP_260901056.1, WP_419970369.1, and WP_420061029.1) exhibited well-defined binding sites with favorable pocket scores (ranging from 0.95 to 53.36), supporting their candidacy for small-molecule targeting (Supplementary Figure 4). Notably, WP_419970369.1 and WP_420061029.1 showed high-confidence pockets with 20 and 41 residues, respectively, and scores exceeding 16, indicating robust drug-binding potential. These results strengthen the case for their inclusion in structure-based drug discovery workflows. However, two proteins (WP_012012715.1 and WP_193220824.1) did not yield any detectable binding pockets in PrankWeb, suggesting that they may not be directly druggable through small-molecule interactions. For this study, we have excluded these two proteins from the final drug target shortlist.
Antigenicity prediction
Antigenicity evaluation of the two predicted vaccine candidate proteins revealed a divergent outcome. WP_260902335.1 scored 0.4304, surpassing the bacterial antigenicity threshold and confirming its vaccine potential. In contrast, WP_004510999.1 yielded a score of 0.3678, falling below the threshold and classifying it as non-antigenic. As antigenicity is a key requirement for immunogenic response, we consider WP_260902335.1 a prioritized vaccine candidate.
Although this study provides comprehensive computational insights into the structure, function, virulence potential, and druggability of HPs in A. butzleri BNI-3166, further experimental validation is essential to substantiate these findings. Future research should aim to perform cloning, heterologous expression, and purification of prioritized proteins for in vitro functional assays to verify enzymatic activity, ligand binding, or interaction specificity. Additionally, gene knockout or mutagenesis experiments in bacterial systems would help determine the role of these proteins in growth, survival, and pathogenesis. Finally, immunological assays and in vivo studies should be performed to assess vaccine potential, while high-throughput screening can identify antimicrobial compounds targeting the predicted novel proteins.
Conclusion
In this study, a comprehensive in silico analysis was performed to explore the functional potential of HPs from A. butzleri BNI-3166. We initially identified 20 ENH proteins as pathogen-specific, of which 10 were functionally annotated with high confidence using multiple domain-based bioinformatics tools. These proteins are predicted to participate in diverse biological roles, including enzymatic activity, binding, transport, regulation, and potential virulence. Subcellular localization analysis suggested that eight proteins—localized to the cytoplasm, periplasm, or inner membrane—may serve as putative drug targets, while two proteins associated with the outer membrane or extracellular space were identified as possible vaccine candidates. Druggability assessment using the ChEMBL database revealed that nine of the ten proteins had no similarity to known drug targets, indicating their novelty and potential for therapeutic exploration. Structural modeling using AlphaFold3 provided insights into their tertiary structures, with predicted pTM scores ranging from 0.44 to 0.92, supporting the feasibility of structure-based drug discovery approaches. To refine these predictions, we performed ligand binding site analysis and antigenicity screening. As a result, six proteins were confirmed to possess well-defined binding pockets and are prioritized as putative drug targets. One protein, WP_260902335.1, was classified as antigenic and retained as a vaccine candidate. The remaining targets were deprioritized due to lack of binding sites or antigenicity. Overall, our findings demonstrate the value of integrative computational pipelines in identifying high-confidence therapeutic and vaccine targets in emerging pathogens. These results provide a solid foundation for future experimental studies aimed at validating these predictions and advancing translational research against A. butzleri.